Did you know you can highlight text to take a note?
x

Suggestions
Use up and down arrows to review and enter to select.Please wait while we process your payment
If you don't see it, please check your spam folder. Sometimes it can end up there.
If you don't see it, please check your spam folder. Sometimes it can end up there.
Please wait while we process your payment
By signing up you agree to our terms and privacy policy.
Don’t have an account? Subscribe now
Create Your Account
Sign up for your FREE 7-day trial
Already have an account? Log in
Your Email
Choose Your Plan
Individual
Group Discount
Save over 50% with a SparkNotes PLUS Annual Plan!
 payment page
payment page
Purchasing SparkNotes PLUS for a group?
Get Annual Plans at a discount when you buy 2 or more!
Price
$24.99 $18.74 /subscription + tax
Subtotal $37.48 + tax
Save 25% on 2-49 accounts
Save 30% on 50-99 accounts
Want 100 or more? Contact us for a customized plan.
payment page
Your Plan
Payment Details
Payment Summary
SparkNotes Plus
You'll be billed after your free trial ends.
7-Day Free Trial
Not Applicable
Renews April 25, 2024 April 18, 2024
Discounts (applied to next billing)
DUE NOW
US $0.00
SNPLUSROCKS20 | 20% Discount
This is not a valid promo code.
Discount Code (one code per order)
SparkNotes PLUS Annual Plan - Group Discount
Qty: 00
SparkNotes Plus subscription is $4.99/month or $24.99/year as selected above. The free trial period is the first 7 days of your subscription. TO CANCEL YOUR SUBSCRIPTION AND AVOID BEING CHARGED, YOU MUST CANCEL BEFORE THE END OF THE FREE TRIAL PERIOD. You may cancel your subscription on your Subscription and Billing page or contact Customer Support at custserv@bn.com. Your subscription will continue automatically once the free trial period is over. Free trial is available to new customers only.
Choose Your Plan
For the next 7 days, you'll have access to awesome PLUS stuff like AP English test prep, No Fear Shakespeare translations and audio, a note-taking tool, personalized dashboard, & much more!
You’ve successfully purchased a group discount. Your group members can use the joining link below to redeem their group membership. You'll also receive an email with the link.
Members will be prompted to log in or create an account to redeem their group membership.
Thanks for creating a SparkNotes account! Continue to start your free trial.
We're sorry, we could not create your account. SparkNotes PLUS is not available in your country. See what countries we’re in.
There was an error creating your account. Please check your payment details and try again.
Please wait while we process your payment
Your PLUS subscription has expired
Please wait while we process your payment
Please wait while we process your payment
Proteins are some of the most interesting and complex molecules in plants and animals. In essence, they have been the entities that have been selected for and against during the course of evolution, their structures and functions shaped and perfected by natural selection. The evolution and mutation of proteins can be realized through changes in deoxyribonucleic acid (DNA), the blueprint for all the proteins that the entire body produces. DNA is translated to proteins via ribonucleic acid (RNA). Although every cell contains an identical copy of DNA with complete instructions for all types of body tissues, only certain proteins are produced by each cell type. In this way, cells of different tissues can perform diverse tasks through the production of unique proteins.
Proteins are composed of long chains of amino acids. Eleven of the twenty amino acids cannot be synthesized by the body and must be obtained through the diet. Since these amino acids are necessary for protein biosyntheses, they are called essential amino acids and include histidine, isoleucine, lysine, methionine, phenylalanine, threonine, tryptophan and valine. The other amino acids can be synthesized by the liver and are called nonessential amino acids.
Each amino acid contains a carboxylic acid group (COOH), an NH2 amino group and
one of twenty functional (R) groups.
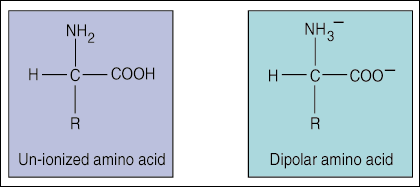
The distinguishing feature of amino acids are their side chains or R groups. R groups can be either acidic, basic, polar or neutral depending on their structure and formula.
Acidic and Basic Amino Acids
Like zwitterions, acidic and basic side chains can ionize depending upon the pH of the surrounding solution. The amino acids that form charged side chains in solution are lysine, arginine, histidine, aspartic acid, and glutamic acid.
Please wait while we process your payment



