
 payment page
payment page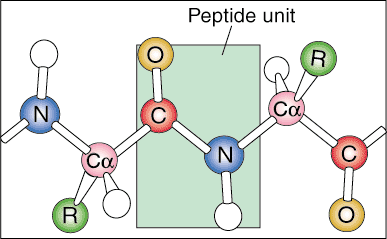
Protein Hierarchy: Primary, Secondary, Tertiary and Quaternary Structure
Proteins have several different levels of organization. They become highly organized and efficient biological machines through many types of ionic and molecular interactions within the protein itself.
Primary Structure
The first level of protein structure is called its primary structure. The primary structure of a protein is simply the linear sequence of its constituent amino acids. Linear sequences are not found in nature because the protein begins to fold as it is produced from messenger RNA.
Secondary Structure
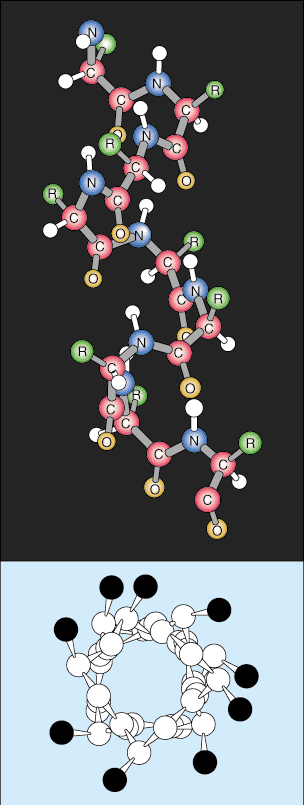
The next level of organization is called the secondary structure of the
protein. The linear sequence of the protein begins to fold into regular
repeating patterns. The two most common secondary structures of proteins are
the alpha helix and the beta sheet.
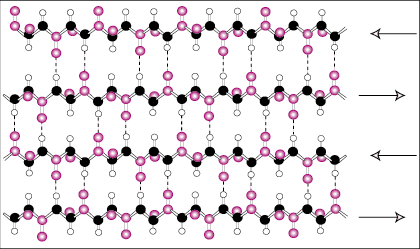
The beta sheet is similar to the alpha helix in that it uses extensive hydrogen
bonding to stabilize itself but it is completely different in structure. The
polypeptide chains are almost completely extended and the hydrogen bonds are
found between different polypeptide chains instead of within the same chain like
the helix.
Tertiary Structure
The next level of organization is called the tertiary structure of the protein. The tertiary arrangement is basically a higher level of protein folding. As the secondary structures become spatially further apart along the polypeptide chain, the polypeptide chains begin to interact with their respective side chains, creating a more complex level of folding. Covalent interactions between cysteine groups, noncovalent dipole-dipole interactions between polar groups, and Van der Waal (induced dipole) interactions between nonpolar R groups are very common in tertiary structures.
Quaternary Structure
Quaternary structure is the last level of protein architecture. Quaternary structure refers to the spatial arrangement of the subunits within the protein. Subunits are categorized as individual polypeptide sequences that begin with a positively charged amino group and end with a negatively charged carboxylic acid terminus. These subunits are formed from individual messenger RNA transcripts and come together to form dimeric (two subunits) or multimeric (more than two subunits) structure. For example, the protein hemoglobin is composed of two pairs of identical subunits that are united by noncovalent interactions.
Protein Folding
How do proteins fold? The complexity of proteins and the number of amino acids involved in folding seemingly create a formidable task. First, most proteins are designed so that their exterior side chains interact favorably with their environment. For example, proteins that are found in water are able to overcome energy barriers required for folding through a process known as hydrophobic collapse. In this process, the hydrophobic or "water fearing" side chains interact more favorably with themselves than with water and use the energy in this reaction to create a hydrophilic exterior and a hydrophobic interior. In contrast, the proteins found in lipid, nonpolar membranes fold in just the opposite manner. The nonpolar residues in the protein face outward, into the membrane while the polar and charged residues face inward to interact with themselves. Many membrane channels and pumps are known to have nonpolar, membrane-spanning amino acid sequences in their structure.
This method of folding sounds very simple; it is not. Although proteins do have machinery to help them fold, proteins must fold through a random search for stable intermediates. Therefore, the protein does not fold all at once. By trial and error, the protein finds most stable intermediates until the final three-dimensional protein configuration is energetically very stable in its environment. With this configuration the protein can maintain its function and structural integrity.
Although the substructures within the protein fold spontaneously, there are so many possible conformations that a protein can adopt that it would take thousands of years for it to assume its proper structure. Yet actual protein folding times are on the order of seconds. The difference between the actual and theoretical times of protein folding is called Levinthal's paradox. It is now known that proteins do not fold through a completely random search but rather take shape through the retention of partially correct intermediates. As more and more of the protein secondary structure folds, the number of possible tertiary structures collapses; as more tertiary folding takes place, the possibilities for quaternary structures similarly decrease. In other words, proteins progressively fold through the stabilization of intermediates rather than by a random search.
Once a protein has folded, it is not invincible. Certain conditions such as temperature and pH can denature a protein. Denatured proteins are proteins that have lost many of their most stable interactions, rendering them inactive or dysfunctional. Since the body acts to maintain a temperature of 37 degrees Celsius and a pH of 7 throughout its tissues, enzymes will function more efficiently in these conditions. If these conditions are disrupted, proteins will begin to denature, disrupting many important tissues including the liver.



