Did you know you can highlight text to take a note?
x

Please wait while we process your payment
If you don't see it, please check your spam folder. Sometimes it can end up there.
If you don't see it, please check your spam folder. Sometimes it can end up there.
Please wait while we process your payment
Get instant, ad-free access to our grade-boosting study tools with a 7-day free trial!
Learn more

![]()

Create Account
This site is protected by reCAPTCHA and the Google Privacy Policy and Terms of Service apply.
Log into your PLUS account
Create Account
Select Plan
Payment Info
Start 7-Day Free Trial!
Select Your Plan
Monthly
$5.99
/month + tax
Annual
$29.99
/year + taxAnnual
2-49 accounts
$22.49/year + tax
50-99 accounts
$20.99/year + tax
Select Quantity
Price per seat
$29.99 $--.--
Subtotal
$-.--
Want 100 or more? Request a customized plan
Monthly
$5.99
/month + taxYou could save over 50%
by choosing an Annual Plan!

Annual
$29.99
/year + taxSAVE OVER 50%
compared to the monthly price!
| Focused-studying | ||
| PLUS Study Tools | ||
| AP® Test Prep PLUS | ||
| My PLUS Activity | ||
Annual
$22.49/month + tax
Save 25%
on 2-49 accounts
Annual
$20.99/month + tax
Save 30%
on 50-99 accounts
| Focused-studying | ||
| PLUS Study Tools | ||
| AP® Test Prep PLUS | ||
| My PLUS Activity | ||
Testimonials from SparkNotes Customers
No Fear provides access to Shakespeare for students who normally couldn’t (or wouldn’t) read his plays. It’s also a very useful tool when trying to explain Shakespeare’s wordplay!
Erika M.
I tutor high school students in a variety of subjects. Having access to the literature translations helps me to stay informed about the various assignments. Your summaries and translations are invaluable.
Kathy B.
Teaching Shakespeare to today's generation can be challenging. No Fear helps a ton with understanding the crux of the text.
Kay H.
Testimonials from SparkNotes Customers
No Fear provides access to Shakespeare for students who normally couldn’t (or wouldn’t) read his plays. It’s also a very useful tool when trying to explain Shakespeare’s wordplay!
Erika M.
I tutor high school students in a variety of subjects. Having access to the literature translations helps me to stay informed about the various assignments. Your summaries and translations are invaluable.
Kathy B.
Teaching Shakespeare to today's generation can be challenging. No Fear helps a ton with understanding the crux of the text.
Kay H.
Create Account
Select Plan
Payment Info
Start 7-Day Free Trial!
Payment Information
You will only be charged after the completion of the 7-day free trial.
If you cancel your account before the free trial is over, you will not be charged.
You will only be charged after the completion of the 7-day free trial. If you cancel your account before the free trial is over, you will not be charged.
Order Summary
Annual
7-day Free Trial
SparkNotes PLUS
$29.99 / year
Annual
Quantity
51
PLUS Group Discount
$29.99 $29.99 / seat
Tax
$0.00
SPARK25
-$1.25
25% Off
Total billed on Nov 7, 2024 after 7-day free trail
$29.99
Total billed
$0.00
Due Today
$0.00
Promo code
This is not a valid promo code
Card Details
By placing your order you agree to our terms of service and privacy policy.
By saving your payment information you allow SparkNotes to charge you for future payments in accordance with their terms.
Powered by stripe
Legal
Google pay.......

Thank You!
Your group members can use the joining link below to redeem their membership. They will be prompted to log into an existing account or to create a new account. All members under 16 will be required to obtain a parent's consent sent via link in an email.Your Child’s Free Trial Starts Now!
Thank you for completing the sign-up process. Your child’s SparkNotes PLUS login credentials are [email] and the associated password. If you have any questions, please visit our help center.Your Free Trial Starts Now!
Please wait while we process your payment
Sorry, you must enter a valid email address
By entering an email, you agree to our privacy policy.
Please wait while we process your payment
Sorry, you must enter a valid email address
By entering an email, you agree to our privacy policy.
Please wait while we process your payment
Your PLUS subscription has expired
Please wait while we process your payment
Please wait while we process your payment
Month
Day
Year
Please read our terms and privacy policy
Please wait while we process your payment
Determining the Rate Law
The goal of a kinetics experiment is to measure the concentration of a species at a particular time during a reaction so that a rate law can be determined. However, it is exceedingly difficult to get an accurate measurement of a concentration at a known time because the techniques used to measure concentrations don't work instantaneously, but rather take time to perform. One of the earliest methods used to measure concentrations at specified times is to quench the reaction either by flash freezing it or by adding a substance that severely inhibits the reaction. Both of these techniques are problematic because one can't be sure that the reaction has completely stopped. The reaction may still be going on during the analysis. Additionally, the reaction mixture is destroyed for the purposes of kinetic experiments, so the chemist must make multiple trial runs and waste a large amount of reagents to observe the concentrations at multiple points in time.
A more modern technique to measure concentration is absorbance spectroscopy. This experiment may be used when a product or reactant has an absorbance frequency unique to those of other components of the reaction mixture. By measuring the absorbance of a particular product or reactant at a variety of known concentrations, you can construct a plot of absorbance versus concentration called a Beer's Law plot. This calibration chart allows you to calculate the unknown concentration given the reaction solution's absorbance. The advantage of this method is that a large number of data points with well known times can be quickly collected using only one reaction mixture.
When looking at the expression for the , you should notice that the variables in the equation are the concentration terms and the powers p and q:
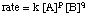
Because we can measure the concentrations in the rate law using the techniques described above, the unknowns we wish to measure are k, p, and q. One method of directly measuring k, p, and q is called the method of initial rates. By measuring the initial rate (the rate near reaction time zero) for a series of reactions with varying concentrations, we can deduce to what power the rate depends on the concentration of each reagent. For example, let's use the method of initial rates to determine the rate law for the following reaction:
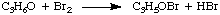
whose rate law has the form:
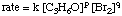
Using the following initial rates data, it is possible to calculate the order of the reaction for both bromine and acetone:
Please wait while we process your payment



