





Basics of Molecular Orbital Theory
Valence Bond theory, as we saw in the last section, is based on the notion that electrons are localized to specific atomic orbitals. Molecular orbital theory asserts that atomic orbitals no longer hold significant meaning after atoms form molecules. Electrons no longer "belong", in a sense, to any particular atom but to the molecule as a whole. Molecular orbital theory holds, as its name suggests, that electrons reside in molecular orbitals that are distributed over the entire molecule.
Quantum mechanics specifies that we can get
molecular
orbitals through a linear combination of atomic orbitals; that is, by
adding and subtracting them. How do we add and subtract orbitals? The best way
to picture this process is to recall the wave-like nature of electrons. Recall
from physics that two waves can interact either through constructive
interference, in which the two waves reinforce each other, and
destructive interference, in which the two waves
cancel each
other out. Mathematically, constructive interference corresponds to addition
and destructive interference corresponds to subtraction. When atomic orbitals
interact, we can either add them to obtain a bonding orbital or subtract
them to obtain an antibonding orbital. Antibonding orbitals are denoted
with an asterisk (*).
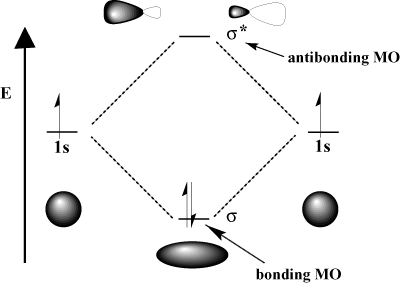
For instance, in the hydrogen molecule, the atomic 1s orbitals can overlap in a σ (head-on) fashion to form a σ-bonding molecular orbital and a σ-antibonding molecular orbital. The bonding molecular orbital is "bonding" in the sense that it is lower in energy than its component atomic orbitals. Forming a bond and moving electrons into the bonding orbital lowers the total energy of the system, which is favorable. On the other hand, moving electrons into the antibonding orbital raises the energy of the system, which disfavors bond formation. The total number of orbitals is conserved; the number of molecular orbitals equals the number of original atomic orbitals.
Polar Bonds
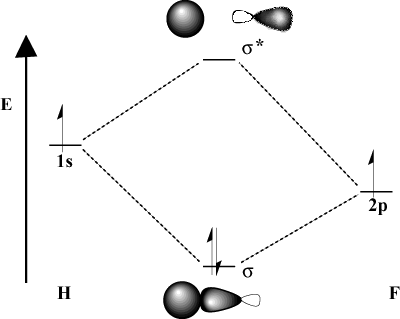
When the bonding atoms significantly differ in electronegativity, their orbital energies will also be different. Compare the MO picture of hydrogen with that of H-F. Because fluorine is more electronegative than hydrogen, its 2p orbitals lie below the 1s orbitals of hydrogen. Due to this difference in energy, the stabilization energy of the resulting bonding MO is not as large. Furthermore, the bonding MO is much more like the 2p orbital in its spatial characteristics while the antibonding MO is much more like the 1s orbital.
Energy Considerations
One advantage of the MO Model is that it gives us more information about the energies of the bonding electrons. In the case of H2, the molecule is stabilized by twice the ΔE of the bonding orbital. The larger this energy gap, the more stable the bond. In order for this stabilization energy to be large, several factors are important:
- The atoms must be of similar electronegativity.
- Atoms must be of comparable size.
- Orbitals must achieve adequate spatial overlap.
Bond Order
How does MO theory describe multiple bonds? In MO theory the bond order of a bond is the number of bonding electron pairs minus the number of antibonding electron pairs. Intuitively, bonding electrons stabilize the bond while antibonding electron destabilize the bond. The greater this difference, the stronger the bond, and the higher the bond order. As we'll see, in MO theory the bond order is no longer limited to integer values.



